Introduction
Pharmacogenomics is an important research field that studies the impact of genetic variation of patients on drug responses, looking for correlations between Single Nucleotide Polymorphisms (SNPs) of the patient genome and drug toxicity or efficacy. The large number of available samples and the high resolution of the instruments allow microarray platforms to produce huge amounts of SNP data. To analyze such data and find correlations in a reasonable time, high-performance computing solutions must be used. Cloud4SNP is a bioinformatics tool, based on the Data Mining Cloud Framework (DMCF), for parallel preprocessing and statistical analysis of SNP pharmacogenomics microarray data.
SNPs
A nucleotide variation or Single Nucleotide Polymorphism (SNP) is defined as a stable substitution of a single base of DNA occurring with a frequency of more than 1% in at least one population.
Pharmacogenomics
Pharmacogenomics is the branch of genomics that studies the impact of individual genetic variations on the response to drugs. In particular, pharmacogenomics correlates gene expression or SNPs with the toxicity or efficacy of a drug, with the aim of improving drug therapies according to the patients’ genotype (e.g., choosing drugs that best match each patient’s genetic profile).
Cloud4SNP: A Cloud Application for Pharmacogenomics
Cloud4SNP is a cloud-DMET-Analyzer version, a bioinformatics tool designed for parallel processing and statistical analysis of Single Nucleotide Polymorphisms (SNPs) data, with a focus on PGx. It efficiently manages large-scale PGx datasets, addressing computational challenges in analyzing genetic variations and drug responses. As a Software-as-a-Service (SaaS) solution, Cloud4SNP eliminates the need for local installations, offering a web-based interface that broadens access to high-performance computing resources. This democratizes PGx research, benefiting smaller research groups, while also introducing challenges related to sensitive data security in healthcare organizations.
Operationally, Cloud4SNP allows to statistically test the significance of the presence of SNPs in two classes of samples using the well known Fisher test. The Fisher test allows to test if two distributions are significantly different, i.e. the eventual difference is not due by chance. In particular, Cloud4SNP performs the following main steps: i) loading of the input dataset and sample class assignment; ii) execution of statistical tests (e.g., Fisher test); and iii) statistical correction of p-values.
The Cloud4SNP Framework
The tool performs several main steps:
- Loading and Assignment: Loading the input dataset (a matrix of alleles) and assigning samples to specific classes (e.g., Class A or B).
- Statistical Testing: Testing the significance of the presence of SNPs using the Fisher test to determine if differences between distributions are not due to chance.
- P-value Correction: Performing multiple tests corrections, such as Bonferroni or False Discovery Rate (FDR), to adjust p-values and correct for false positives.
Three new tools have been added in the Data Mining Cloud Framework to support parallel processing of SNP input data: i) a Partitioner tool, which creates a set of partitions from a single SNP dataset; ii) a ModelMerger tool, which merges into a single model the partial models generated by the DMETAnalyzer, either corrected or not by a Corrector; iii) a ModelsMerger tool, which takes in input three single models (with FDR, Bonferroni or none correction) and produces a single HTML file. Using the web GUI of Data Mining Cloud Framework, the tools described above have been composed into the Cloud4SNP workflow shown in Figure 1.
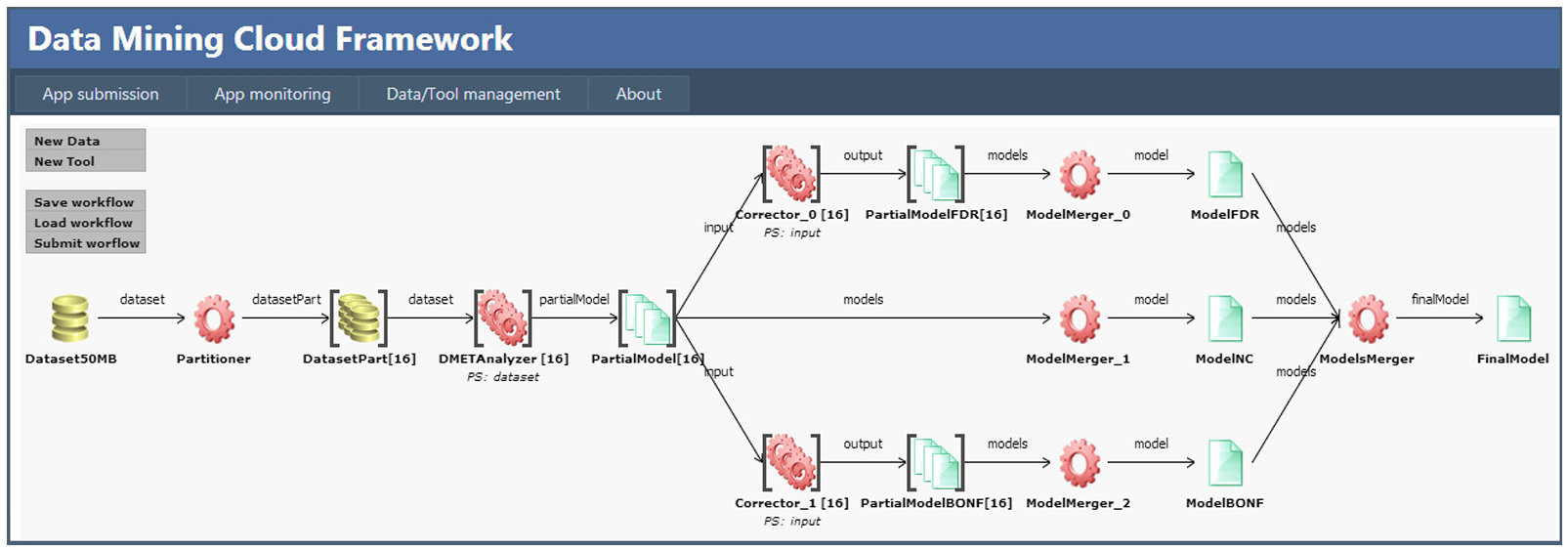 Fig. 1. Cloud4SNP workflow in the Data Mining Cloud Framework.
Fig. 1. Cloud4SNP workflow in the Data Mining Cloud Framework.
The workflow performs the following steps. As first step, the initial dataset is partitioned into n chunks by the Partitioner, where n is the number of workers available for parallel data processing (16 in this case). By increasing the number of workers, it is possible to improve the level of parallelism and, therefore, system performance. Each data chunk DatasetPart[i], i = 1,…,n, is analyzed by an instance of the DMETAnalyzer tool (i.e., DMETAnalyzer[i]), which produces a partial model (i.e., PartialModel[i]) containing the p-values of each probe. Such partial models are corrected using two different instances of the Corrector tool: Corrector 0 that uses an FDR correction, and Corrector 1 that uses a Bonferroni correction. Then, three instances of the ModelMerger tool are used to create three models, ModelNC, ModelFDR and ModelBONF, which are respectively the model with no corrections (composition of PartialModel[n]), the model with FDR correction (composition of PartialModelFDR[n]), and the model with Bonferroni correction (composition of PartialModelBONF[n]). Finally, the ModelsMerger tool combines ModelNC, ModelFDR and ModelBONF to produce a single HTML file with all the output results (see Figure 2).
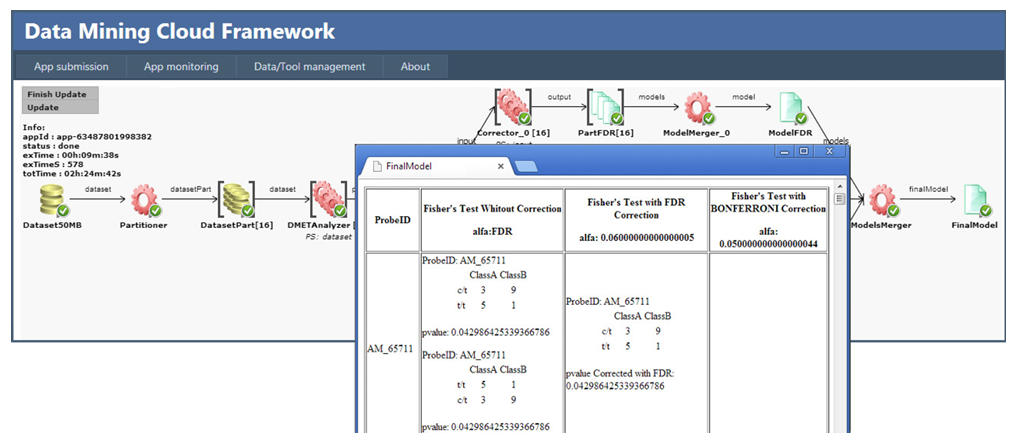 Fig. 2. Cloud4SNP workflow at the end of the execution, with visualization of the final result.
Fig. 2. Cloud4SNP workflow at the end of the execution, with visualization of the final result.
Using Apache Spark for faster in-memory processing
Cloud4SNP has been extended to execute applications on Apache Spark, which provides faster, in-memory executions for iterative and batch processing. Differently from other systems where intermediate data are stored in distributed file systems, Spark stores data in RAM memory and queries it repeatedly so as to obtain better performance. Since the preprocessing, analysis, and statistical correction algorithms have been entirely rewritten in Spark, Cloud4SNP is able to exploit its high-performance features, obtaining faster execution times and a high level of scalability, with a global speedup that is very close to linear values. Experimental evaluation shows that Spark ensures a reduction in the processing times by an average of 35% compared to the other systems.
References
Marozzo F, Belcastro L. High-Performance Framework to Analyze Microarray Data. Methods Mol Biol. 2022;2401:13-27. doi: 10.1007/978-1-0716-1839-4_2. PMID: 34902119.